中南大学阳华/陈凯OL: 光诱导EDA复合物介导的自由基-自由基交叉偶联实现吲哚的C2-苄位C(sp³)–H烷基化
摘要:中南大学化学化工学院阳华/陈凯课题组报道了一种利用光诱导电子供体-受体(EDA)复合物介导的自由基-自由基交叉偶联,以多种2-甲基吲哚类化合物和溴化物为原料,成功实现了吲哚C2-苄位C(sp³)–H的直接烷基化。相关研究成果近期发表在Org. Lett.

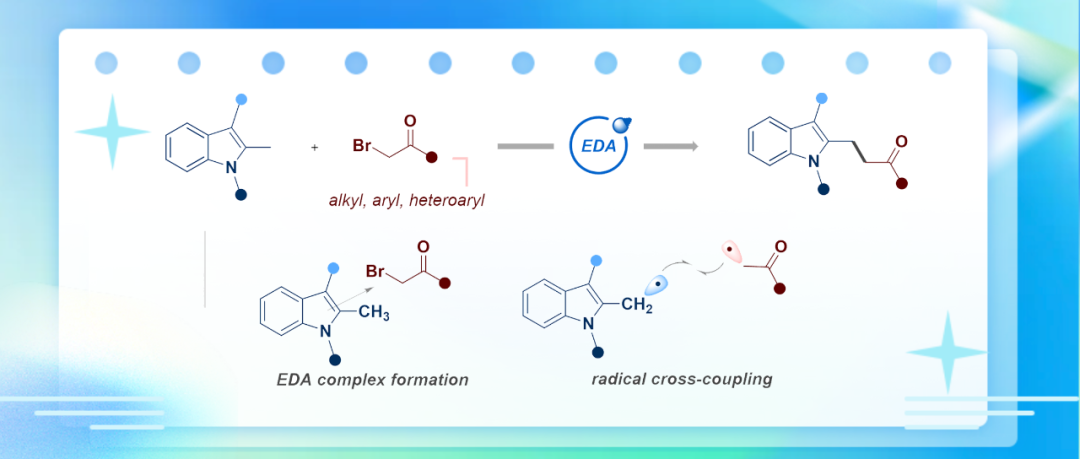
吲哚骨架广泛存在于天然产物、药物分子和功能材料中,其C2位官能化衍生物因其广泛的生物活性而备受关注。传统C2-烷基化策略通常依赖于吲哚富电子特性的Friedel-Crafts反应或需要过渡金属催化、预装导向基团及强酸/碱条件,在步骤经济性和官能团兼容性方面存在局限。近日,中南大学化学化工学院阳华/陈凯课题组报道了一种利用光诱导电子供体-受体(EDA)复合物介导的自由基-自由基交叉偶联,以多种2-甲基吲哚类化合物和溴化物为原料,成功实现了吲哚C2-苄位C(sp³)–H的直接烷基化。相关研究成果近期发表在Org. Lett.(DOI: 10.1021/acs.orglett.5c03141)。
吲哚及其衍生物是一类重要的含氮杂环化合物,其中C2-烷基化吲哚衍生物因其具有广泛的生物活性并作为多功能的构建单元和有价值的合成中间体而受到特别关注(图1a)。当前吲哚C2-苄位C(sp³)–H烷基化的策略通常涉及过渡金属催化、预安装活化/导向基团、或使用强酸或强碱的闭壳层途径(图1b)。相比之下,通过生成C2-苄位自由基物种来揭示吲哚自由基阳离子的新反应性,无疑为多样性导向的C–H键官能化提供了广阔的化学空间。近年来,采用电子供体-受体(EDA)复合物、无需外源光催化剂的可见光介导有机转化受到了显著关注。该课题组基于其在光催化领域的研究基础,合理推断利用富电子的吲哚(电子供体)与缺电子的卤化物(电子受体)可形成EDA复合物。该复合物在可见光照射下发生单电子转移(SET),生成C2-苄位自由基与烷基自由基,最终通过极性匹配的自由基-自由基交叉偶联实现C2α-烷基化(图1c, 1d)。
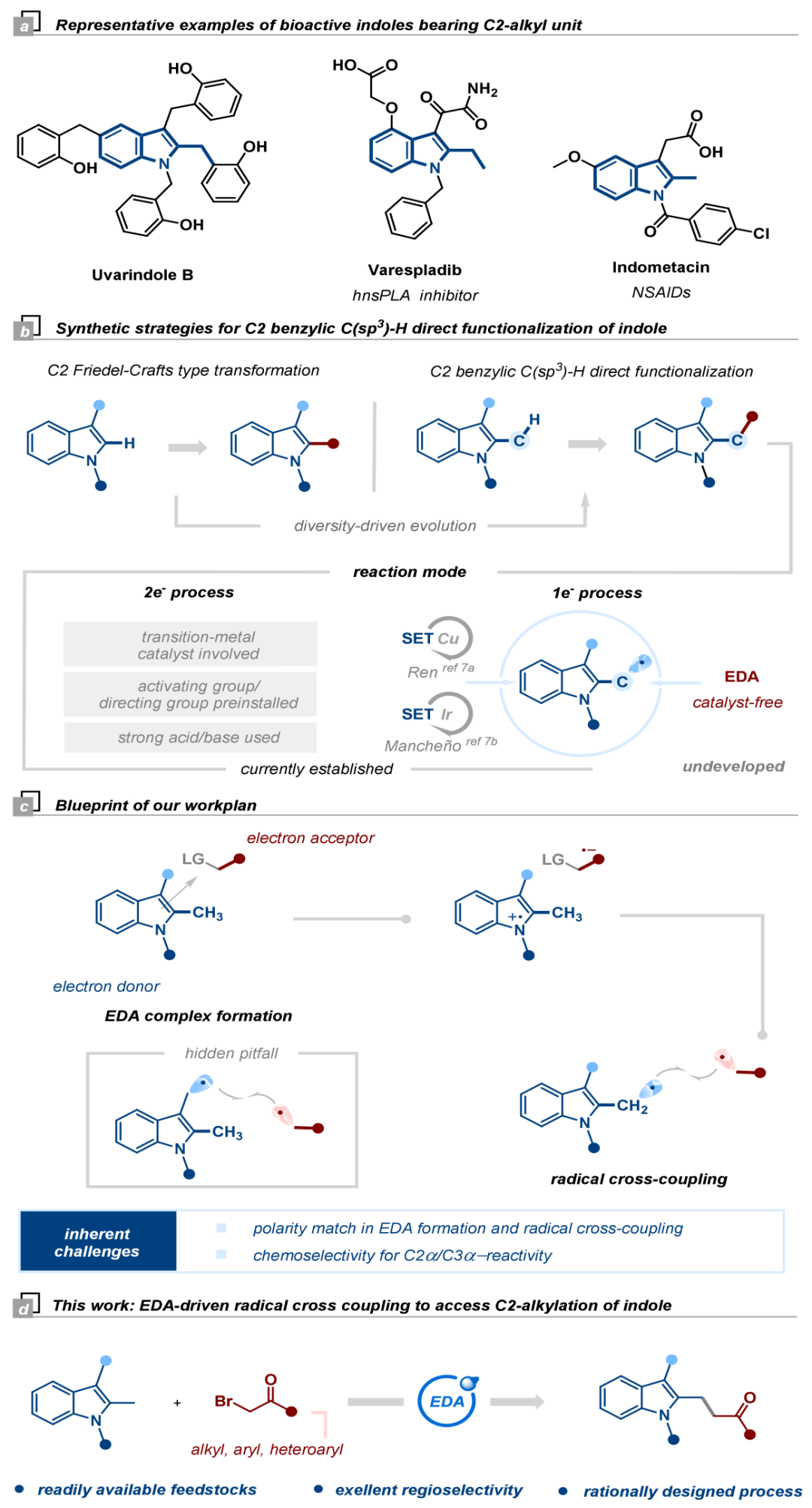
图1.研究背景及本课题的设计思路(来源:Org. Lett.)
首先,作者以1,2,3-三甲基吲哚(1a)和2-溴苯乙酮(2a)为模型底物进行反应条件优化。最终确定最佳条件为:1a(4.0 equiv.),2a(1.0 equiv.),碱K₂CO₃(4.0 equiv.)在溶剂DCE中,于氩气氛围下,经30 W 450 nm LED照射12小时。该条件以71%的NMR产率和65%的分离产率得到目标产物3。反应无需外加光催化剂,且对照实验表明碱和光照对于反应至关重要(图2)。
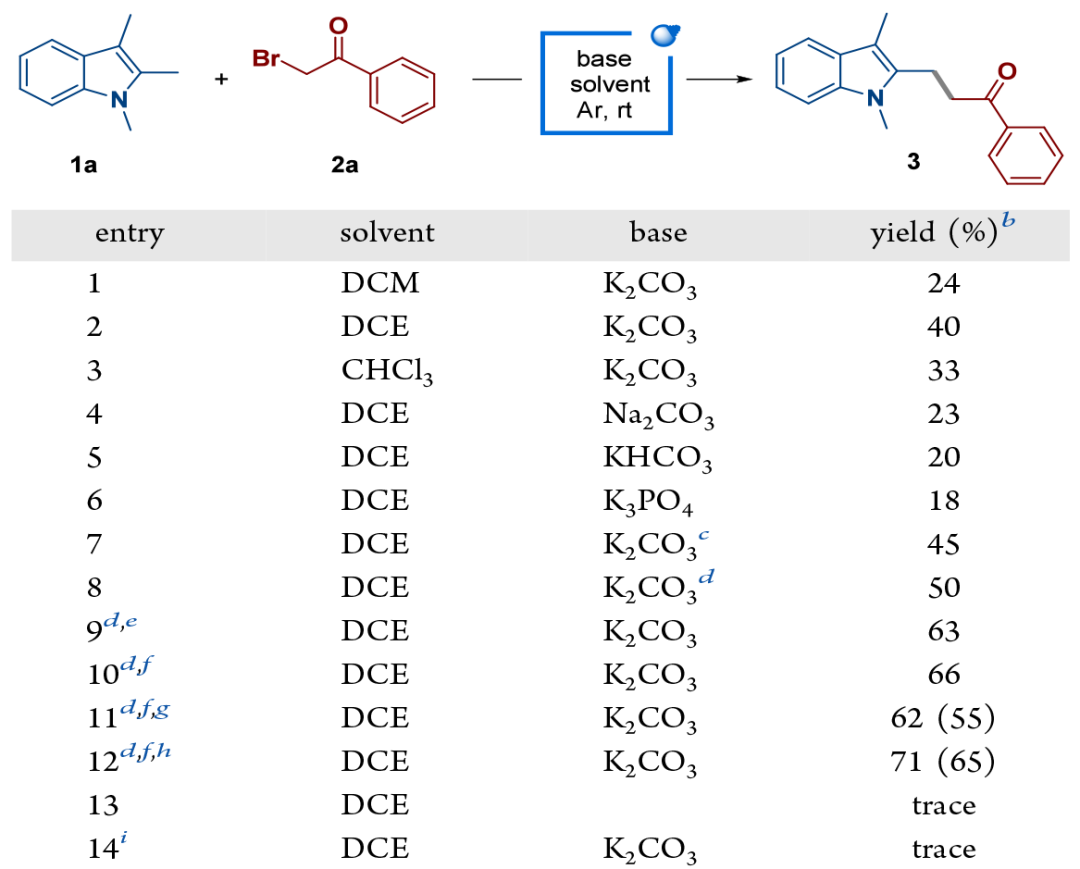
图2.条件筛选(来源:Org. Lett.)
在最优条件下,作者首先对α-溴代酮的适应性进行了探索(图3)。结果表明,苯环对位带有给电子(如甲基、甲氧基)或吸电子(如氯、溴、三氟甲基)取代基的底物均能以良好产率得到相应产物。邻位或间位氟取代底物也能以中等以上产率反应。萘环、噻吩和脂肪族α-溴代酮也是合适的底物,尽管产率相对较低。该策略还可应用于产生仲碳自由基和叔碳自由基的溴化物,其中带有两个吸电子基团的溴代丙二酸二乙酯产率可达65%。值得注意的是,衍生自天然合成香料(如吐纳麝香、萨利麝香)的溴化物也成功参与了反应。
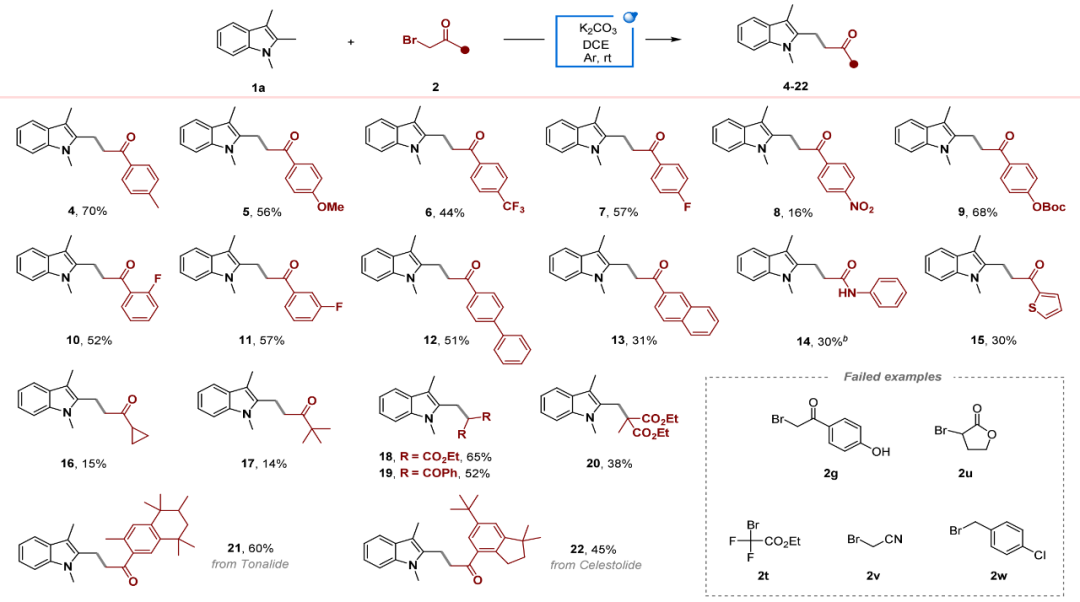
图3.溴代物的底物范围(来源:Org. Lett.)
接下来,作者进一步对吲哚的底物范围进行了考察(图4)。结果表明,N-取代(乙基、苄基、烯丙基、苯基)的2-甲基吲哚都能顺利反应,以中等产率得到目标产物。N-Boc保护吲哚(1n)因降低吲哚核的电子云密度而抑制了反应,N-H吲哚(1o)和四氢咔唑(1p)为底物时,反应体系较为复杂未能检测到目标产物。1,2-二甲基吲哚(1q)意外地得到了C3–H官能化产物。苯环不同位置带有给电子或吸电子取代基的2-甲基吲哚底物均能兼容,以中等产率得到产物。2-乙基吲哚和3-乙基吲哚也可发生反应,并表现出较好的区域选择性(rr值分别为5:1和3:1)。
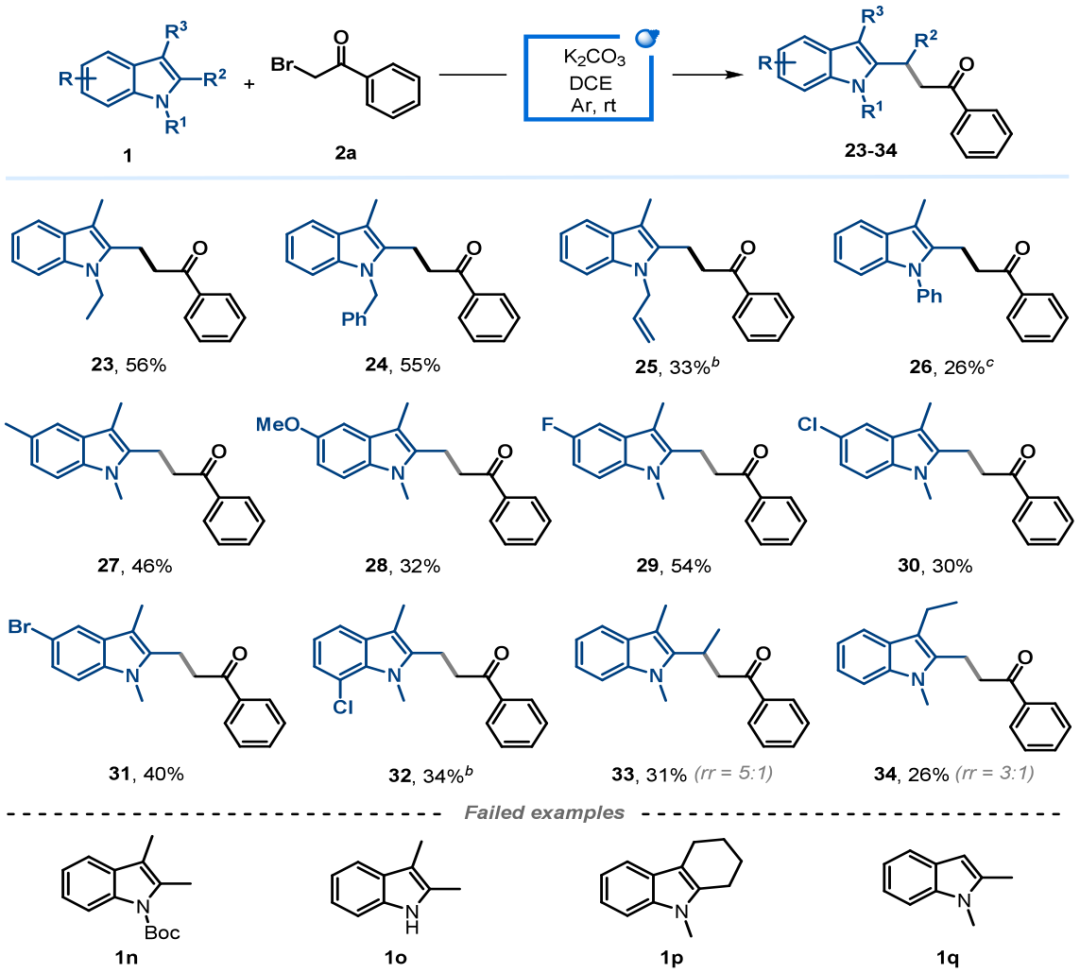
图4.吲哚的底物范围(来源:Org. Lett.)
为进一步验证该合成方法的实用价值,作者开展了放大实验,克级规模反应以略低的分离产率获得产物3(图5)。此外,进行了多样的合成衍生化实验,羰基被NaBH₄还原为醇(35);与PhMgCl发生格氏反应得到叔醇(36);与2-氨基-3-吡啶甲醛缩合构建杂环(37);经SeO₂氧化得到烯酮(38)。
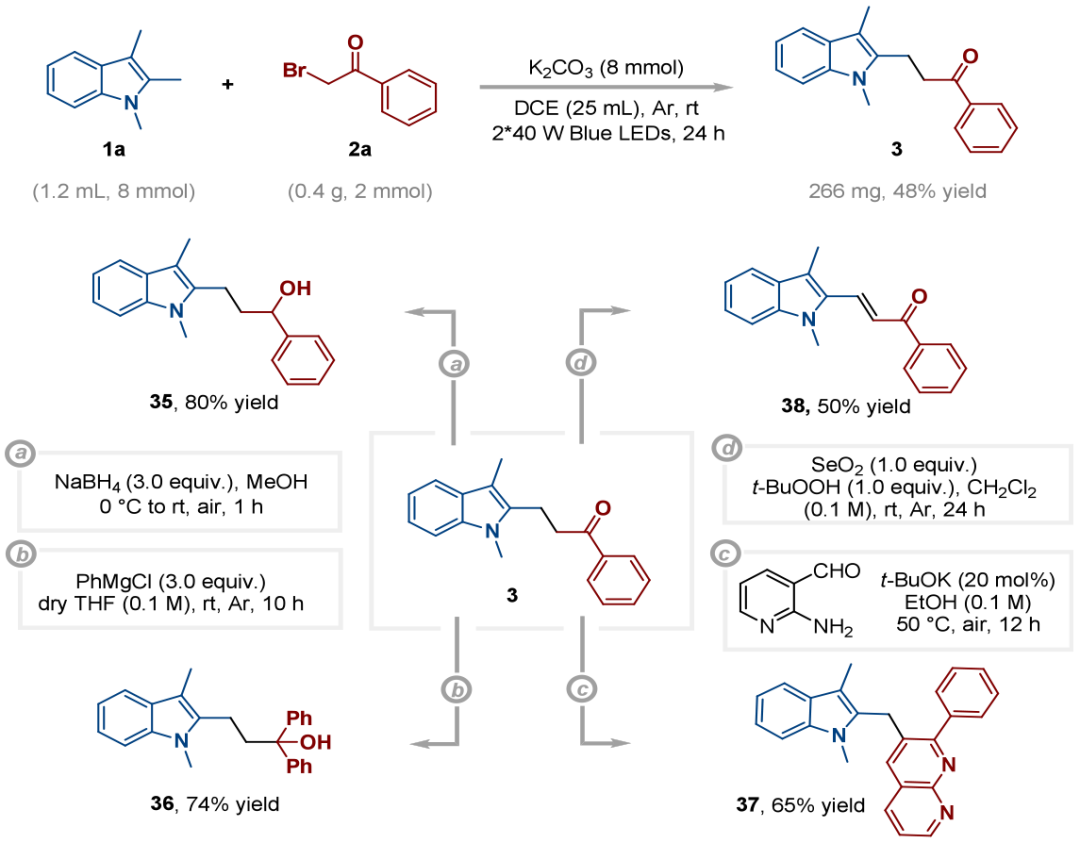
图5.放大合成和衍生化实验(来源:Org. Lett.)
为了进一步探究反应机理,作者开展了一系列控制实验研究(图6)。首先,UV-Vis光谱显示了1a和2a混合后的红移现象,这表明1a和2a 之间可能形成EDA复合物。其次,使用2,2,6,6-四甲基哌啶氧化物(TEMPO)为自由基抑制剂进行自由基捕获实验,大大抑制了产物生成,并通过NMR, ESI-HRMS检测到了相应的自由基加合物,支持了自由基途径。接着,开/关灯实验表明反应依赖持续光照,排除了反应为自由基链式机制的可能性。最后,Stern-Volmer实验表明2a能有效淬灭1a的荧光,表明从激发态吲哚(1a*)到2a的单电子转移(SET)也是可行的。基于以上实验结果,作者提出了可能的反应机理(图6e):EDA复合物A光诱导发生SET,或者1a直接光激发后促进2a的还原裂解,生成吲哚自由基阳离子B和溴化物自由基阴离子C;C裂解产生烷基自由基E和溴离子;吲哚自由基阳离子B在碱的作用下脱质子得到C2-苄位自由基D,最后,C2-苄位自由基D与烷基自由基E发生交叉偶联得到产物3。
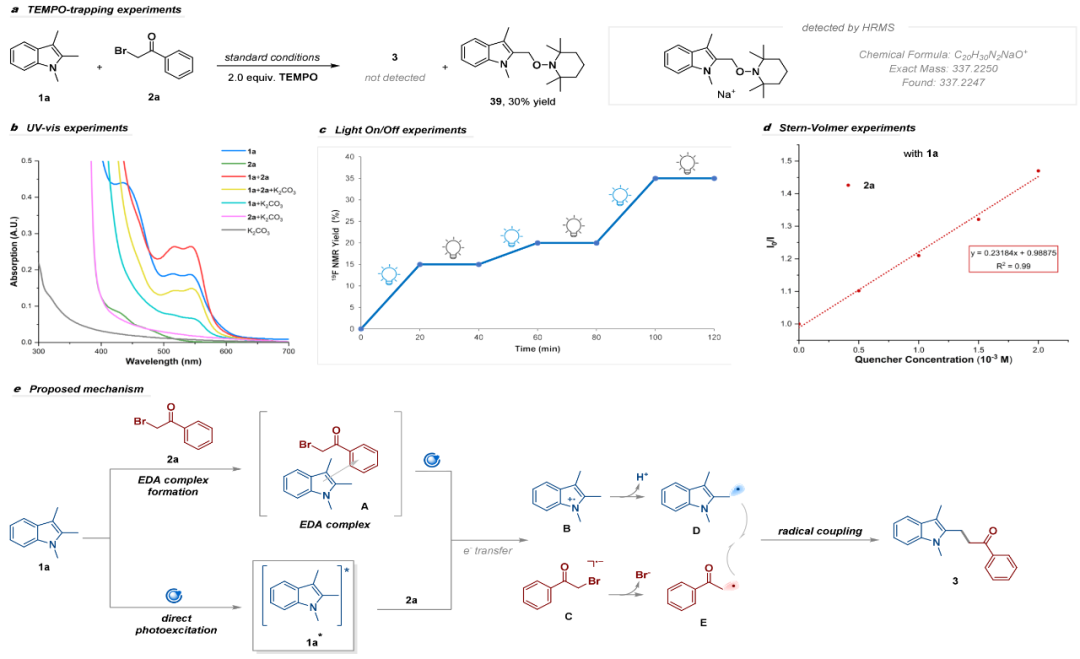
图6.机理研究和可能的反应机理(来源:Org. Lett.)
总之,阳华/陈凯课题组开发了一种新颖、高效的可见光诱导策略,实现了吲哚C2-苄位C(sp³)–H的直接烷基化。该方法的成功关键在于吲哚与溴化物形成EDA复合物以及后续极性匹配的自由基交叉偶联过程。该方法无需外源光催化剂、条件温和、操作简单、底物范围广泛、官能团耐受性好,为结构多样化的2-烷基吲哚库的构建提供了高效、便捷的途径,也为发展新型光化学模式、推动重要杂环化合物的多样性功能化研究提供了新思路。
中南大学化学化工学院阳华教授和陈凯副教授为论文共同通讯作者,中南大学2024级硕士研究生胡盈盈同学和2023级博士研究生刘志林同学为论文共同第一作者。(论文作者:Ying-Ying Hu, Zhi-Lin Liu, Hao-Yue Xiang, Xiao-Qing Chen, Kai Chen,* Hua Yang*)。该研究工作得到了国家自然科学基金、湖南省自然科学基金以及中南大学研究生自主探索创新项目等的资助。
声明:本文仅用于学术文章转载分享,不做盈利使用,如有侵权,请及时联系小编删除。
查看更多
相关阅读
热门文章
热门标签
光催化苯甲醚
光催化自芬顿降解水中有机污染物
釜式光反应器厂商
常州光化学反应仪价格
磺胺甲恶唑抗生素
可见区光化学反应仪
光催化还原技术
光化学有机反应
光催化实验
光催化合成醚类化合物
高通量光反应仪报价
光催化甲烷氧化制甲基
实验室光化学反应釜工作原理
光催化自由基串联环化反应
石英光化学反应釜
光催化有机合成发应
光诱导烷基胺与伯醇的无受体脱氢偶联构建α-氨基酮
光化学反应釜如何选择
多功能平行光化学反应仪
芝加哥大学
光流体微通道反应器小试
光催化析氢设备
光催化反应釜光源
光催化合成苯并噻唑衍
光催化醛与羰基或亚胺的不对称还原交叉偶联
有机合成
光催化降解有机污染物
可见光催化醛的不对称还原炔丙基化反应
有机光光合成过氧化氢
LED单工位光反应仪
紫外光化学反应仪使用说明
可见光催化穿梭二溴化
光化学反应仪哪家好
可见光催化葡萄糖产HMF
烯丙基sp3 C–H键烷基化
洛桑联邦理工学院
盘管连续流反应器
光催化光异构化反应
高压光反应釜应用场景
水光催化去芳构化[2+2]环加成反应
去芳香性戊烯基化反应
光诱导下偶氮官能化合成吲唑
光促进3-烷基吡啶苄基C–H 键自由基氯化
华东理工大学
西湖大学
光催化反应器
光催化烯烃全氟异丙基化反应
光促进吡啶重排构建3-吡啶醇
光驱动三元催化脂肪胺γ-C−H键转化
光诱导铜催化脱羧 C-C偶联
定制光流体微通道反应器
北京光化学反应仪
光催化烯烃的芳基烷基化反应
光化学制备烯烃
光催化炔烃
光催化合成镇痛药芬太尼
光催化反应器用途
光加成反应
光化学介导饱和杂环酮脱氢
有机光催化剂功能区别
光催化制乙烷
光催化烯烃α-酰化反应
光催化制乙烯
光催化氧化糠醛
有机光化学反应原理
多位光化学反应仪厂家
光催化异氰与炔烃的碳/氢氰化反应
光诱导下脂肪烷烃与烯烃的偶联反应
聚四氟乙烯反应釜
光流体微通道反应器量产
光氧催化led光源
可见光催化苄基三级C–H键直接羧基化反应
烯烃的光氧化
光催化环丙烷的去消旋化
光催化产氢
光诱导炔烃双官能团化反应
光催化还原反应
光催化糖类转化制备5-羟甲基糠醛
光催化降解诺氟沙星
光反应釜分类
光诱导烯烃氮杂环丙烷化反应
光催化制备维生素B
光催化乙炔氢氯化反应方法
光催化合成β-酮一级氯化物
光催化有机卤胺烷基化反应
平行光化学反应仪器
重氮化合物可见光诱导反应
香港大学
光催化间苯二酚-甲醛树脂
光催化合成α‑叔伯胺
光化学反应仪搅拌方式
光催化氧化还原
实验室光化学反应仪选购指南
溴二氟烷基
ghx光化学反应仪
光反应仪器选型
光聚合反应
光催化偶联合成烯丙基三级烷基胺
光催化合成三氟甲基酮
光催化烯烃
急需光化学反应仪
上海光化学反应仪厂家
光酶选择性自由基环化
光催化合成芳胺
光诱导合成CF3
光催化芬顿降解
光催化N-酰胺的α-三氟甲硫基化
偶氮苯
光催化氧化5-羟甲基糠醛
二芳基二氢吩嗪基多孔有机聚合物
可见光诱导有机膦促进磺酰肟盐的连续脱氧
微通道反应器透光材质
光氧化还原催化构建C-苷
芳香族异硫氰酸酯
喹啉
工业级光化学反应仪
光催化苄胺和烷基芳烃的C(sp3)−H芳基化反应
溴烷基化反应
光反应釜
光诱导活性聚合物网络
光化学反应釜
光催化构建构建硫杂环丁烷
同济大学
光催化水分解制氢
光化学反应仪温度控制系统
可见光催化合成苯酚
光化学反应仪光学系统
平行光光源
光流体微反应器厂家
光催化解聚木质素
光催化硝基芳烃去芳化扩环合成多取代氮杂环庚烷
光催化烯烃2σ+2π环加成
有机光化学合成策略
N-杂环卡宾非均相光催化
光催化[2+2]环加成反应
光诱导电荷存储
光催化硫醚
微反应器
光催化制醇类化合物
光催化异氰C-N键断裂生成烷基自由基
黄素光催化去饱和与环氧反应
光催化反应釜使用说明书
光催化甲烷氧化制甲醇
玻璃光反应釜温度控制
光催化烯烃的氧炔基化
光催化甲烷制乙烯
光化学反应仪规格
光催化微塑料升级转化偶联
有机
恒温循环水控温的工作原理
光催化合成β2,2-氨基酯
微通道光反应器设计原理
光催化去除水中邻苯二甲酸酯
光催化羧酸盐生成磺酰亚胺酰胺
光催化氧化甲苯制苯甲醛
单光源光反应仪
光催化CH4制C2H4
光催化配套设备
烯烃二卤化
维生素B光合成设备
光化学C-N交叉偶联
光催化硝基芳烃
光催化草酸盐促进醇的脱氧羧酸化
风冷光催化反应器
可见光催化硫氰化反应
光催化交叉亲电偶联反应
光反应仪独立调光
光催化反应速率
小试级光化学反应仪
光催化水处理
光催化合成α-酮酯
四川大学
芳香烯烃高效氧化裂解
光催化合成反应方程式
光降解聚乙烯
多光源光化学反应仪厂家
光化学反应仪光源
可见光微通道反应器
连续流微通道反应器
光催化制备5-羟甲基糠醛
光化学反应釜技术参数
小试级光催化反应器
地屈孕酮分子式
光催化烯烃与三氟甲烷亚磺酸钠发生三氟甲基硫代化反应
微通道光反应器
光氧化还原催化丙烯酸酯
光催化烷烃
可见光催化构建苯并环丁醇
光催化脱羧卤磺酰化反应
光催化降解塑料
光催化脱羧Giese反应
甲苯光催化氧化制苯甲醛
烯烃的氢二氟甲基化
光催化去除有机污染物
内置搅拌光反应仪
插烯反应
光化学反应仪怎么选择
国产光化学反应仪
LED工业光反应器
光催化降解VOCs
光溴化反应
模板法制光催化剂
高压光反应釜
光催化析氢实验装置
光催化BCBs酰胺的烷基芳基化反应
光催化构建酰基缩醛衍生物
光环化反应
光化学反应仪报价
光催化合成多环邻氨基醇类化合物
光催化羧酸化合物结构重塑
光催化微流体反应器设备
光催化构建芳杂胺
搅拌式光化学反应仪
烯烃的连二磺酰化修饰
浙江大学
光降解抗生素
光催化C-C键偶联反应
多功能光反应仪
光催化矿化产物 CO2 转化为 CO
光化学反应仪哪个品牌好
光催化α-羟基酸合成α-酮酸
光氧化合成原理
光流体微通道反应器
光化学反应仪技术标准
可见光催化伯胺类化合物
加州理工大学
清华大学
光诱导催化烯丙基C-H键酰氧基化
光化学反应仪选型
光催化环氧乙烷的开环不对称炔基化反应
玻璃光催化反应釜技术参数
光催化胺化
单孔位全波段光反应仪
多位光化学反应仪报价
哈尔滨工业大学
光催化制备乙醇胺
光催化合成手性双环己烷类化合物
光催化合成BCP醚类衍生物
溴代烷烃
光诱导构建1、2-芳基杂芳基乙烷
光催化led光源
光催化烷烃C–H键固SO2构建砜
光催化降解CIP
光催化甲烷偶联制乙烷
紫外光连续流反应器
光催化饱和杂环酮与胺的脱氢去饱和偶联
g-C3N4
光氧化还原催化芳香醚氢解
光催化氧化胺偶联
光诱导含偶氮苯聚合物可逆固-液转变
邻氟磺酰硼化反应
山东大学
光反应釜容量选型
光催化合成氨基酯
光化学反应釜类型
光催化微流体反应设备
光催化氧化还原反应制备香料中间体
光催化剂降解诺氟沙星
酶启发配位聚合物超分子酸构筑
光催化喹啉衍生物
可见光催化脂肪烃
小试光反应器
光偶联反应方程式
光催化合成轴手性N-芳基吡咯
多位光化学反应仪优势
光氯化反应釜
封端-糖基化
光化学耦合
光催化CO2还原为CH4
光化学合成N-糖苷
光催化芳香醇
光催化反应仪器选型
光催化合成酮类化合物
光催化sp3–sp3氧化偶联立体选择性合成氨基酸
光反应釜搅拌速度
光化学反应釜分类
光催化反应釜功能
光催化丁二烯
光催化脂肪族羧酸脱羧卤代
微通道光催化反应器
光诱导加成实现螺桨烷双官能团化反应
光铜共催化的自由基脱羧偶联反应
烯醇硅醚转化为α,β-环氧酮
高低温光催化反应器
中国科学技术大学
多光源光化学反应仪
烷烃的光氧化
光催化电子转移反应
光催化聚乙烯
光催化构建烯丙基N,O-酰基-缩醛
可见光催化羰基类化合物
光催化氯化苄自偶联反应
实验室多功能光化学反应仪
光催化有机合成地屈孕酮
光催化烯烃与羧酸的氢-氟烷基化反应
光催化降解反应釜
光化学反应仪工作原理
光催化降解挥发性有机物
光催化生物质脱氢
科研级光化学反应仪
光引发的环化反应
有机光化学反应仪
光催化硝酮
光催化炔烃生产伯醇
中国矿业大学
光催化乙烷
光诱导环丙酰胺与炔烃环化构建吡啶
光化学反应仪led光源
光催化合成甲酸苯酯类化合物
光氯化反应釜厂家
生产级盘管式连续流光反应器
光催化有机磷酸双自由基交叉偶联
光子自旋轨道耦合
led光化学反应釜
光催化不饱和烃
光催化构建氮杂环丁烷
6孔平行光反应仪
南京理工大学
光催化反应方程式
天津光化学反应仪报价
单工位全波段光反应仪器
光催化甘氨酸酯α-C–H官能团化构建非天然氨基酸
10孔光化学反应仪
南京光化学反应仪
光催化合成N-烷基苯胺
光催化亲核氟化
ghx光化学反应仪优势
光诱导下烷基胺的远程溴化修饰新方法
国内光化学反应仪
光氧化反应
可见光催化二氯化
海南光化学反应仪
液冷光催化反应器
光电共催化
光催化构建环丙烷化合物
光诱导下硝基氧化合成
光催化降解抗生素
光敏化合成
气相沉积法制光催化剂
6工位光化学反应仪
光氧化还原催化苄基叔碳C-H键与CO2的羧酸化反应
光催化C-H键活化构建α-手性烷基膦
光催化选择性氧化芳香醇
光催化降解VOCs废气
光催化微通道反应器
光催化烯烃芳化氨甲酰化反应
光催化甲烷氧化偶联
光催化去消旋化反应
可见光驱动的单金属交叉亲电偶联反应
光化学反应仪功能
光流体微通道反应器厂家
光催化烯烃烯丙位C-H键与芳基磺酰化反应
光催化氧化硫醇制备二硫醚
光催化一级杂芳基胺
X射线光化学反应仪
光解对四溴双酚A
邻苯二甲酰亚胺自由基
光诱导水促进芳基烯烃C=C的歧化裂解
超分子光-酶偶联催化水污染物绿色降解
高效光催化氧化偶联反应
光催化还原制烯烃
光催化CH4和CO2偶联制乙醇
光催化醇类化合物
光催化烯醇硅醚的α-磺酰化反应
光催化反应釜选型
光催化有机污染物降解
光诱导下EDA复合物多组分交叉偶联新方法
光催化反应器装置
光催化CO₂还原生成C₂烃类化合物
光催化反应釜组成
连续流光反应器设计原理
光诱导脱硫交叉偶联
大容量光化学反应仪厂家
光氧化
福州大学
光反应釜用途
可见光催化硫醇的选择性氧化脱氢偶联
光化学反应仪选购
光环化反应原理
高通量光化学反应仪
光催化制氢
光催化硅基羧酸化合物
光诱导共轭合成1,2,4-三氮唑
光催化合成C4化合物
光催化氮类化合物化学转化
高精密光化学反应仪
光催化技术
光催化炔烃合成N-芳基吡咯类化合物
光催化甲苯
光催化过氧化氢
紫外光化学反应仪波长
光微通道反应器
紫外光反应器
光催化domino反应构建硫杂环丁烷
光反应仪统一调光
光催化喹啉衍生物C(sp2)-H官能化反应
光氧化还原催化苄位选择性酰化反应
光化学反应仪优势
石英通道连续流反应器
光催化基本原理
光诱导下SO2固化合环修饰
光催化C-H键活化机理
光化学反应釜选型
光催化CO2环加成反应
光催化环己酮脱酰芳基化
多工位光化学反应仪
光诱导下串联合环修饰新方法
光催化剂合成原理
光电催化醇的C-C键断裂转化
光功能材料制备
光催化脱氧氢烷基化修饰
光催化还原六价铬
红外区光化学反应仪
可见光诱导的钴催化烯烃双膦化反应
光催化合成磺酰亚胺酰胺
光功能材料表征方法
可见光催化还原硝基化合物
光催化降解五氯苯酚
光催化合成维生素B
光氯化反应釜定制
天津大学雷圣宾教授课题组
光诱导烯烃的插氮合环氮杂环丙烷化转化
光催化制乙醇
金属笼光催化制氢
led光催化反应釜
工业级光催化降解反应釜
光催化合成高炔丙醇化合物
光催化环丙酰胺无受体脱氢开环
有机光化学基本概念
led光反应仪
光催化助力烯丙基C-H键胺化
宾夕法尼亚大学
光催化制α‑叔伯胺
光致异构化合成药物设计
光催化氯二氟乙酸与烯烃的多样官能团化反应
光催化氧化法
齐齐哈尔大学
酮烯胺
可见光驱动硫脲和羧酸选择性合成
微通道反应器作用
光化学反应仪解决方案
光催化还原二氧化碳制乙烷
光催化Meerwein型溴芳基化反应
筒式微通道反应器
光催化合成稠多环邻氨基醇类化合物
光催化反应器工作原理
光催化烯烃合成β-氨基酸衍生物
光催化苯基甘氨酸
光环化反应方程式
水制氢
光催化析氢装置
光化学合成多环邻氨基醇类化合物
可见光催化氯化反应
光催化生物质多元醇制备乙二胺
可见光诱导下氮杂尿嘧啶的C−H硅基化反应
平行光反应仪选型
光催化氧化降解盐酸四环素
叔烷基胺
光催化反应器香料合成
光催化合成钛磺酸框架
光化学促进环己酮脱氢去饱和C-N偶联
光引发的重排反应
复合光催化剂
光催化合成2-羟基苯并呋喃-3(2H)-酮
可见光催化杂环C-H烷基化反应
连续流光催化反应器
光化学反应仪波长
光诱导电荷转移复合物
微通道反应器光催化制地屈孕酮
光化学反应仪应用
光催化反应釜医药合成
光催化降解四环素
多通道光化学反应仪
南方科技大学舒伟
光催化剂种类
光催化环丙烷胺酰化反应
光化学反应仪多少钱
光谱学技术药物研发
光催化乙苯
有机光催化剂应用领域
开放式光催化光源功率
光反应釜选型
纳米光催化剂
光催化羧酸
光催化剂合成
光催化脱羟糖自由基的N-糖基化反应
会聚对电解-光催化策略
光催化合成噁唑
高精密光化学反应仪报价
玻璃光反应釜压力
朴玲钰
光催化合成α-氨基膦酸酯
光化学反应仪市场
光催化氢氟磺酰化
光催化串联实现[3+2]环化构建α-SCF3环戊酮
光催化还原反应釜
平行光反应仪特点
光催化构建轴手性N-芳基喹唑啉酮
光催化降解甲胺
光诱导交叉偶联
光催化合成合成环状胺
前景
光氧化还原共催化烯烃氨酰化合成β2,2-氨基酯
光催化苯甲醚酰胺化
光诱导下双核Au催化偕二氯代化合物多样化硼化修饰
全波段光催化反应器
光降解反应釜应用场景
光催化芳基烯烃C=C键的歧化裂解
光催化合成芳基-烷基硫醚
烯烃环异构化合成杂环
光化学反应仪光源选择
聚合物
光催化偶联反应
光诱导实现天然糖类化合物位点选择性
光催化合成氮杂环丁烷
单孔光化学反应仪
维生素B分子式
光催化合成β- 氨基酯
光诱导下芳基卤代物与羧酸化合物合成硫酯
基于芳基噻蒽鎓盐的烯烃
光催化药物降解
光催化合成松香烷型二萜
光连续流反应器
南开大学
光催化烯烃的溴烷基化反应
连续流光反应器制备异噻唑
有机光反应器
光催化的不对称烯烃异构化
光降解刚果红染料
光催化BCP醚类衍生物合成
光催化环己烷脱氢
加州大学伯克利分校
柱式微通道反应器
可见光催化芳香杂环氮自由基
光催化降解PAEs
二苯乙烯光异构化
光催化BCBs烷基芳基化反应
石英微通道光反应器压力设计
光反应釜容量
光催化气态烷烃和芳基溴化物偶联
常州光化学反应仪
光催化CO2还原
光催化呋喃与胺的亲核-亲核偶联反应
光氧化还原-NHC
半导体光催化剂
石英微通道反应器
不锈钢反应釜
光催化苯胺
石英微通道光反应器
光催化卤代反应
红外光化学反应仪
光催化反应釜作用
光催化C−H键杂芳化反应
光诱导Pd催化丁二烯与吲哚的串联不对称去芳构化反应
光化学反应仪led光源功率
XPA光化学反应仪
光化学反应仪维修
光芬顿法
光催化合成氮杂芳环羧基化合物
马来酰亚胺
光氧化还原双催化
自由基加成反应
有机光化学合成基本原理
可见光催化烯烃双官能团化反应
光催化脱氢还原
光化学反应仪进口
光催化自由基极性翻转环加成策略合成环状胺
光催化丙酮
生产级连续流光反应器
光异构化反应
光催化合成α-氨基膦氧
三甲基氯硅烷
光合成甲醇
郑州光化学反应仪厂家
光催化微流体反应装置
乙烯和CO₂合成可光降解聚乙烯
高压光化学反应釜
光诱导下芳烃C-H 键直接胺化反应
紫外光反应仪
烯烃
偕溴代硝基环丁烷
光催化剂选择
可见光合成多取代吡咯
光化学反应仪哪家性价比高
光催化Meerwein型溴芳
光催化生成烷基自由基
光催化苯甲醇
光诱导构建γ-硫代内酰胺
光催化乙炔制备氯乙烯
光催化甲烷无氧偶联制乙烯
光催化亚磺酰胺
可见光催化有机硫去氯氢反应
光化学反应
光催化烯烃自由基还原交叉偶联
南京光化学反应仪哪家好
板式微通道光反应器
高压光反应釜压力设计
有机光催化剂分类
光催化不对称还原交叉偶联
邻苯二甲酸酯的碳和氢同位素分馏
光催化香料合成反应
有机化合物脱氢偶联
光催化降解聚对苯二甲酸乙二醇酯塑料
光催化光源波长选型
光催化制备硫醇
光促铜催化脱羧卤磺酰化反应
LED光催化反应器应用
光催化交叉偶联构建烯丙胺
光催化CO促进羰基化
光催化合成光学活性联烯
光诱导吡啶扩环
大连工业大学
光催化C-N偶联耦合
光催化氧化降解抗生素
可见光催化从环己酮
EDA 复合物
光催化诱导BCPs三组分自由基接力反应
华南理工大学
科研级光催化反应器
光催化轻质烷烃与芳基溴的偶联
光催化降解水中四环素类抗生素
光化学反应仪转速
光催化原位生成芳基磺铵盐
南京光化学反应仪选哪个品牌
郑州大学
光催化反应釜材质
光催化实现C-杂原子与富电子芳基偶联
光催化合成吲哚酮类化合物
光诱导芳基三氮烯与CDCl3的氘化反应
光催化促进吡啶C4-选择性氟烷基化
光催化合成2-哌啶酮
光催化碳原子删除的色原酮二烯骨架编辑
光催化实现烯烃的芳氮化
多相光催化氧化降解抗生素
光催化三氯甲基烯烃内酯化
玻璃光反应釜
光化学合成有机化合物
光催化诱导脂肪胺α-C(sp3)−H键膦酰化
光催化还原重金属离子
绿光照射下酮的 α 芳基化的光氧化
光还原反应
光催化苄胺偶联
光催化水消毒
光催化末端烯烃
光催化乙酸偶联生产丁二酸
光催化反应釜
光催化自由基脱羧偶联反应
自由基
微通道反应器工作原理
可见光催化合成酰胺和N-酰基脲
光催化降解沙星类抗生素
光催化生物质多元醇制备乙醇胺
光催化乙烷氧气脱氢制乙烯
光催化制氢反应仪
光催化降解氧氟沙星
光催化降解仪器
合成可光降解聚乙烯
光催化生成C₂烃类化合物
光电催化芳烃的脱羧三氟甲基化
可见光诱导合成杂芳基碳-糖苷
氮化硼
实验室光化学反应釜
对硝基苯光催化还原制苯胺
可见光光催化芳烃的C−H胺化反应
分子光谱学原理
甲烷氧化偶联制轻烷烃
光催化硫醚的选择性氧化
光反应釜功能作用
光氯代反应
单孔光反应仪
不锈钢微通道反应器
上海多试管光化学反应仪
光反应仪
武汉光化学反应仪价格
可见光诱导肟酯C-C键官能化反应
光催化降解氯酚类化合物
光催化反应釜维护保养
郑州光化学反应仪
常温平行光反应仪
光化学反应优势
C-H键活化偶联
光催化合成E-烯丙醇
光催化羧酸与芳基碘的酯化反应
可见光催化剂耦合高级氧化工艺
光催化合成维生素B工艺流程
广东工业大学
水热法制光催化剂
光催化烷氧羰基保护仲胺的α-烷基化
光化学合成基本原理
光催化乙烯基酮共轭加成
光化学反应仪保养
实验室光反应釜
β-内酰胺
光催化BCB自由基阳离子的[2π + 2σ]环加成反应
光诱导不对称还原交叉偶联
光催化加成反应
多试管光化学反应仪多少钱
10工位光催化反应器
光催化反应釜操作指南
光反应釜组成
室温光催化氧化甲烷
光化学反应仪常见问题
光催化合成醇类化合物
光催化降解装置
光催化降解亚甲基蓝
光催化反马氏氢胺化反应
异相光催化氯化苄自偶联
分子光谱学概念
光催化丙酮偶联制备2,5-己二酮
高通量光反应仪工作原理
光化学反应仪用途
光催化制氢设备
光催化烯丙基C–H酰氧基化反应
光电催化二氧化碳还原产甲醇
善施科技完成 Pre-A 轮融资
光催化5-羟甲基糠醛
有机光催化剂应用场景
光化学合成连二醇
光催化苯胺和DIPEA的偶联反应
液冷光化学反应仪
光催化芳基氯化物与醇类合成硫醚
光合成生物学
光催化二甲醚
光催化亚磺酰自由基与烯烃加成
深紫外光化学微反应器
光反应釜设计温度
光氧化还原吡哆醛自由基生物
光催化构建α-SCF2H环戊酮
光酶选择性控制自由基反应
光催化串联反应
深紫外光化学反应仪
光催化合成多官能团34-二氢喹唑烷酮
光催化剂制备原理
光催化羧酸与S8的脱羧C-S构建
光催化有机合成钛磺酸框架
多试管光化学反应仪价格
微通道反应器持液量设计
平行光反应仪厂家排名
光化学反应仪维护
光反应釜材质
可见光催化还原炔丙基化反应
单工位光反应仪器
可见光诱导构建非天然氨基酸
开普敦大学
光流体微反应器报价
康奈尔大学
高压光反应釜
玻璃光催化反应釜
光催化医药合成反应釜
偶联
光化学led光源
风冷光化学反应仪
光催化Heck类偶联反应
光化学反应釜报价
光催化还原CO2制乙烷
光加成反应原理
三氟甲基硫代化反应
光催化卡宾与腈
芳烃的光氧化
光功能材料基本原理
水凝胶
光催化氧化衍生醇
等离子体催化
高压光反应釜耐压
武汉光化学反应仪
光催化合成β-氨基酸酯
光催化合成
光催化医药合成反应仪
光催化农药合成
光化学合成β-氨基酸酯
西安交通大学
光化学反应仪恒温循环水控温
光催化[3+2]环加成反应
光催化合成含硼杂环化合物
光诱导芳烃分子内环加成去芳构化
海南光化学反应仪价格
光电催化苯乙烯碳胺化
平行光化学反应仪
光催化伯胺类化合物
平行光反应仪实际应用
可见光诱导肟酯C-C键的断裂
光化学反应仪内置搅拌原理
光催化合成苯甲腈
光催化对四溴双酚A
光流体微通道反应器参数
紫外可见光化学反应仪
合成气和烯丙基sp3 C–H键的选择性芳基化/烷基化
光催化合成苯并咔唑类化合物
光催化氧化氮氧化物
玻璃光化学反应釜
胺的直接C-H官能化
光催化合成手性烯丙基砜类化合物
光催化烯烃烷氧基重氮甲基化
光化学反应仪采购
微通道反应设备
有机光催化剂表征方法
光催化合成乙烷
光催化合成磺酰胺
光诱导催化构建Z-烯烃
光催化合成磺酰氟化合物
光催化制备乙二胺
精密光化学反应仪
第23届有机合成国际会议
光催化合成亚磺酰胺
北京理工大学
光催化烯烃不对称双官能团化
光化学反应仪时间设置
有机光催化剂
光催化实现烯烃与醇的脱氧交叉偶联
多试管光化学反应仪
高通量光催化反应器
光催化多组分不对称Minisci反应构建手性β-卡波林
光催化缺电子吲哚衍生物
光催化胺烷基化构建α‑叔伯胺
光催化降解邻苯二甲酸酯
光催化酰胺自由基插入双环丁烷转化
三氯甲基化反应
光反应器点光源
光催化合成甲醇
光催化N-苯基哌啶的去饱和 β-C(sp3)–H酰胺化方法
光诱导紫苏迷迭香酸生物合成
光催化析氢耦合苯甲醇氧化
光引发的自由基反应
光催化合成氟烷基酮化合物
阿德莱德大学
光催化析氢有机耦联反应
光催化丁二烯的不对称胺化
实验室平行光反应仪
OER
光催化酰胺BCBs与α-羰基烷基溴的自由基加成反应
Science
光催化烯丙基C-H键胺化构建支链胺
光催化色酮
可见光驱动有机转化
石英板式微通道光反应器
亚甲基蓝
光催化合成合成烯丙基硼
光催化聚乙烯转化为乙烯
单工位光化学反应仪
光催化合成超高分子量聚合物
光催化聚乙烯转化为乙烯和丙酸
盘管式聚合物微通道光反应器
光催化苯甲醚类化合物
光催化还原CO2
光催化苯甲醚酰胺化反应
组合式反应釜
光催化构建芳基烷烃
光催化α-羟基酸合成酒石酸衍生物
CO2还原
光催化酰基三氮唑构建酮
光催化合成硫醚
光化学反应过程
光催化芳基卤
光化学反应仪产物分析
UV光化学反应仪
光化学反应仪波波长选择
LED光化学反应仪
光催化1,2-氨基醇合成
光加成反应方程式
光诱导多组分分子内环化/羟基三氟甲基化级联反应
光化学合成策略
光化学反应仪行业标准
溴代氟酰基芳烃与多种烯烃的自由基环化反应
水相硫-芳基抗体偶联
光催化烷基溴三氟乙酰化反应
光诱导下脱氧官能化修饰新方法
聚合物微通道反应器
固定床光反应器厂家
马来酰亚胺衍生物
磁力搅拌器
光催化非活化烯丙醇的半频哪醇重排反应
光催化降解盐酸四环素
光催化C-N偶联耦合产氢
水裂解
开放式光源
硝基芳烃
苄胺底物光催化偶联
光催化氧化反应
复旦大学
光催化降解技术
光催化甲苯选择性氧化
光化学反应仪在医药领域的应用
光化学反应仪哪家比较靠谱
光催化还原制苯胺
多功能光催化反应器
光催化修饰香料分子
光催化合成氨基酸衍生物
光催化富电子芳烃的碳-杂原子偶联
微通道连续流光反应器
光催化制二芳基硅化物
微通道反应器设计参数
光催化制备地屈孕酮
光催化合成芳胺及衍生物
中国地质大学
光诱导草酸生成二氧化碳自由基阴离子
平行光反应仪厂家
微通道反应器
铀酰光催化烯烃氧化裂解
光诱导烯基重氮[4+2]环加成反应
光催化脱羧反应
高通量光反应仪厂家
光化学反应仪器厂家
光催化合成酯类化合物
光引发偶联反应
单工位光催化反应器
光致异构化合成原理
光催化降解设备
光氧化还原双重催化
光降解反应釜
光催化合成2,3-二胺
风冷控温6孔光反应仪
玻璃通道光催化反应器
连续流光反应釜
多电子
光自由基加成反应
光催化去除四环素
平行光反应仪精准液冷控温
有机磺酸
光催化制酮
可见光催化氧化烷烃
光催化降解有机物
光化学合成应用
高温高压光反应釜
光催化醛-烯烃偶联
单工位LED光反应器
光去羰基反应
光催化亚磺酰胺实现烯烃的胺芳基化
CO2光催化还原
光催化烷基卤的C-N偶联反应
光催化甲醇脱氢
光诱导吡啶结构的骨架编辑
光催化聚对苯二甲酸乙二醇酯瓶子公斤级降解
可见光催化降解黄药
有机光化学反应应用
光催化反应釜厂家
光催化还原法
光催化交流研讨会
光催化烯烃和一级杂芳基胺的反马氏氢胺化反应
光催化降解酚类物质
光催化烯烃的氢化四氟异丙基化
光催化一级杂芳基胺与烯烃的分子间反马氏氢胺化反应
光催化磺胺嘧啶
光催化构建支链烷基胺
温控光化学反应仪
光催化制合成三氟甲基酮
氨基悬链烯烃
催化羧酸不同自由基的C-C交叉偶联
固定床光反应器选型
三氟甲基化
光催化剂实现单电子转移
光催化醛-烯偶联反应
科研级光反应器
光催化促进亚磺酰胺合成
光氧化还原双重催化唑类化合物与芳基碘的C–H芳基化反应
光催化环丙叔醇
有机光催化剂基本原理
光催化水分解产氢
光促进羧酸与S8的脱羧C-S构建
光还原反应方程式
光催化氧化
光催化降解聚乙烯
光催化降解水中污染物
光催化构建手性β-卡波林
有机光化学合成应用
平行光反应仪光源
光促进杂环苄基C(sp3)–H键多样性转化
光催化合成α-氨基羰基化合物
大通量光化学反应仪
光催化α-氨基自由基与烯烃加成
光化学反应釜价格
光化学反应仪光源分类
光氧化还原
噻蒽鎓盐光催化
光化学反应仪贵吗
光化学反应仪厂家
led光反应仪器
甲烷
单孔位光反应仪
光催化丁二烯与吲哚的串联不对称去芳构化反应
光化学反应仪厂家排名
光氧化还原协同催化丙二烯的芳磺酰化
石英光反应釜
光催化硝基苯加氢反应
华南师范大学
平行光反应仪品牌
光催化醛不对称形式交叉偶联
平行光催化反应器
光催化合成H2O2
可见光还原交叉偶联
光催化反应釜光柱
光催化构建环状内酯衍生物
多位平行反应釜
科研级光反应仪器
上海光化学反应仪厂家排名
光催化反应釜降解
光催化硫醚化反应
不锈钢光反应釜
光催化塑料降解
光催化甲烷选择性氧化制甲醇
光诱导碘代烷烃的胺化反应方法学
电催化制备硒基噁唑酮衍生物
双供体-受体有机网络光合成过氧化氢
釜式光反应器选型
光自由基加成反应原理
光催化合成原理
可见光催化芳香烯烃氧化裂解
多相光催化氧化降解TMP
光催化氧化糠醛制备四氯化碳化合物
烷基炔烃的双官能化转化
大容量光化学反应仪价格
光流体微通道反应器定制
光还原催化乙烯和CO₂合成可光降解聚乙烯
光催化NHP酯合成手性烯丙基胺衍生物
光催化胺与羧酸构建亚磺酰胺
光催化合成烷基氟磺酰化合物
光化学反应仪器生产厂家
有机光催化
微通道反应装置
光催化反应器在有机合成领域的应用
工业级盘管式连续流光反应器
光催化制维生素D
光诱导下Co催化下Semipinacol重排新方法
光化学反应仪维护保养
丙酮光催化脱氢偶联制备2,5-己二酮
平行光反应仪光源参数
光催化偶联
光催化降解甲硝唑
实验室光催化反应器
led光催化反应设备
光催化氧化甲烷制甲醇
内置光催化光源
流动化学光反应器
光催化C-H官能团化实现吲哚的不对称去芳构化
光化学反应釜厂家
微通道连续流反应器在医药生产中的应用
光催化耦合类芬顿反应去除污染物
光催化芳烃对位选择性C-H键胺化新策略
有机光催化剂制备
光催化水氧化
光催化CO₂还原制C2H4
光催化α-C–H键芳基化
光流体微通道反应器工作原理
光催化芳烃直接胺化
光催化合成过氧化氢
光催化生成乙烯
全波段光反应仪
光催化芳香醚氢解
光化学合成芳基胺
丙烯酰苯胺自由基环化反应
全波段光反应器
常温光化学反应仪
光催化制高炔丙醇化合物
多位光化学反应仪特点
光催化烯烃双三氟甲硫基化反应
釜式光反应器
光催化
光催化苯甲酸酯与烯丙醇的脱氧交叉偶联
光催化合成地屈孕酮
光催化芳基环丙烷
高精密光化学反应仪厂家
光催化烯丙基醋酸酯
光流体微反应器多少钱
微通道光反应器分类
深紫外光催化反应器
开放式光催化光源
光催化水净化器
光催化环加成反应合成手性双环己烷类化合物
光催化甲基酮脱酰炔基化反应
光催化药物合成
光催化CO2还原制备C2H4
光化学反应仪设备
二硫化物光化学合成
光化学反应仪市场分布
上海光化学反应仪报价
地屈孕酮光催化设备
多相光催化
可见光催化芳基环丙烷开环官能团化反应
光催化去除难降解的芳香族污染物
光催化烯丙醇的半频哪醇重排反应
led光化学反应仪控温
可见光催化苯
光催化合成芳基胺
光生电荷
UV光反应器波长
实验平行光反应仪
光催化氧化制甲醇
光催化制备四氯化碳化合物
全波段LED光反应仪
光催化制醇
重庆大学
氟聚合物
平行光反应仪保养
香港城市大学
光催化木质素解聚
有机光化学反应类型
氢烯基化
连续流反应器
光催化反应光源
紫外单孔光反应仪
光催化糖类
光催化环丙烷
Angew
光催化制甲醇
光流体微通道反应器中试
近红外区光化学反应仪
光催化氧化技术
光催化聚乙烯转化为丙酸
光催化微流体反应器
光催化合成维生素B反应方程式
钙钛矿光催化剂
光催化二芳基醚分子转化为两个苯酚分子
光氧化还原催化实现脱羧交叉偶联
光诱导催化邻烷基硝基苯的吖啶化转化
光化学合成仪
光化学反应仪说明书
光化学合成地屈孕酮
烯丙基sp3 C–H键的选择性芳基化
光诱导H键-EDA复合物促进烯烃氢化硫化
有机光催化合成2-哌啶酮
光诱导
华中师范大学
光催化末端烯烃生产伯醇
光催化降解环丙沙星
100ml微通道光反应器
光偶联反应
光催化低温乙苯转化
光催化实现醛的α-叔烷基化
光催化构建富含 C(sp3)的偕二硼砌块
光催化合成反应仪
光诱导烷基羧酸化合物的脱羧
光催化合成苯甲酸
连续流光化学合成
可见光催化
光化学基本概念
光诱导芳基碳碘键活化
光催化分解反应
光催化降解芳香族污染物
光氧化合成应用
光化学合成
光催化三氟甲烷亚磺酸钠
微通道反应器压力值设计
有机光化学反应机理
连续流光反应器分类
光催化应用
有机光催化合成地屈孕酮
微通道连续流反应器
水光催化轻烯烃的双羟基化制备二元醇
布里斯托大学
光催化α-叔碳伯胺合成
光催化析氢
光催化反应釜反应腔
光催化双分子烯烃还原偶联
新加坡国立大学
光催化木质素模型化合物
光催化连续流反应器
溶胶凝胶法
光催化气体烷烃与芳基溴的交叉偶联
光催化降解水体有机污染物
光催化芳构化驱动C-C键断裂的交叉偶联
光反应釜报价
光催化三氟乙酸对芳烃的三氟甲基化
光催化反应器类型
光催化芳基羧酸脱羧氧化
液冷控温光化学反应仪
清洁可再生能源化学合成
UV光反应器
光催化反应原理
甲烷光催化氧化偶联
多光源光化学反应仪报价
紫外光催化反应器
光催化生成2-吲哚酮
光催化环加成反应
光催化原理
可见光催化苯和脂肪烃选择性偶联
光化学反应仪功率
光催化吡啶骨架编辑构建双环吡唑啉
东京大学
光诱导烯烃化学选择性芳杂化
光催化剂设计
二维富勒烯
玻璃光催化反应釜工作原理
光催化制备环丁烷
光催化羧酸盐生成亚磺酰胺
光催化反应釜冷却装置
光催化构建α-CF3-炔
光氧化合成
光谱学技术有机化合物鉴定
光催化制氨基酸
光诱导脱羧烷基化反应
羧酸
光化学反应仪
邻苯二甲酰亚胺
光催化5-羟甲基糠醛转化2,5-二甲酰基呋喃
光化学合成氮杂环庚烷
LED光催化反应器
盘管式连续流光反应器
光催化反应釜工作原理
气固相光催化反应器
叠氮化物
光化学反应仪控温方式
实验室光催化反应釜
光催化烯烃的胺-磺酰胺化修饰
光催化降解法
厦门大学
微通道反应器压力值
光催化反应器常见故障问题
光催化甲烷氧化
光催化偶联制乙醇
溶胶凝胶法合成光催化剂
可见光催化合成腈类化合物
光催化杂环苄位C-H键氯化
光诱导合环构建环丁烷
光催化构建氟取代叔脂肪高烯丙基胺
光诱导催化选择性脱羧二氟甲基化
氮杂环丙烷
光化学硝基苯去芳构化
非均相光催化
光催化产业化
光催化二甲苯氧化反应
微通道反应器技术参数
光催化降解苯酚
可见光催化合成手性醇类化合物
固定床光反应器优势
单工位光反应仪
烯烃双官能团化反应
烯二炔的环化
光催化烯烃的氨基-羧基化反应
光氯代反应原理
光催化有机合成
光催化甲烷制乙醇
光诱导下偕二氯化合物环丙烷化
光化学反应仪选哪家
光催化反应机理
硼化
Au-CeO2
光化学反应仪波波长
国内光化学反应仪现状
光还原反应原理
光氧化硫醇-烯交叉偶联反应
光催化水相硫-芳基抗体偶联
玻璃微通道反应器
糠醛
光催化制醚
光催化制备醛
光催化木质素生物质转化为芳香族单体
光诱导LMCT脱羧
钍簇
金属氧化物光催化剂
连续流微通道光反应器
有机合成光化学反应仪
光化学反应仪实验结果
光致异构化合成
可见光光源
实验室光化学反应仪
光催化唑类N-H与烯烃的氢胺化反应
光催化合成酰基酮产物
光催化降解PET-12塑料
可见光催化醛和铵盐合成腈类化合物
大连理工大学
光催化甲烷转化制备乙醇
光催化降解抗生素废水
led光反应釜
led光化学反应仪波长
可见光催化脱羧溴化反应
玻璃通道连续流反应器
光催化制地屈孕酮
光催化降解二氯苯酚
光催化烯烃的芳硅化反应
C–H双官能团化反应
天津光化学反应仪厂家
北京大学
海南光化学反应仪厂家
烷基磺酰化反应
光催化甲苯制苯甲醛
光诱导下三元催化下γ-氨基的官能化转化
盘管式光催化反应器
光催化三氟乙酰化反应
加州大学洛杉矶分校
光催化甲烷制乙酸
光催化烯烃α-三氟甲基化
光催化反应器使用注意事项
光氧化还原反应
光催化LED光源
6孔平行光化学反应仪
可见光催化螺碳环化合物的解构氟化反应
光化学反应仪LED光源
紫外光化学反应仪多少钱
光催化制备氨基酸
光催化aza Paternò–Büchi反应构建氮杂环丁烷
光催化水分解
光诱导脱氢偶联
光化学反应仪使用说明书
光催化构建全季碳羧酸化合物
光化学反应仪使用指南
贵州大学
光催化碳氟磺酰化
剑桥大学
光催化构建α-氨基酸
紫外光化学反应釜
光催化降解对四溴双酚A
可见光诱导膦催化的烯烃自由基环化
多相光催化氧化降解废水中抗生素
多试管光化学反应仪厂家
光偶联反应原理
光催化CO促进杂芳基迁移反应
光催化合成环丁烷氧化吲哚骨架
光催化合成氮杂环庚烷
光催化香料合成反应仪
光催化氧化反应釜
可见光催化呋喃转化成吡咯化合物
光催化环烷烃与苄溴的C−C键交叉偶联
led光化学反应仪
固定床光反应器
光催化氧化二氧化硫
光催化降解磺胺甲恶唑抗生素
光催化卡宾自由基阴离子插入反应
不锈钢光化学反应釜
光诱导的扁桃酸与醇氧化酯化反应
紫外光化学反应仪厂家
光催化降解家禽粪便厌氧消化液中有机污染物
光催化分解水制氢
10工位光化学反应仪
光催化芳基氯与醇合成芳基烷基硫醚
室温光催化氧化甲烷制备液相产物
光化学反应仪精度
光催化领域
光催化酰胺化反应
光催化聚合反应
磺胺嘧啶
武汉大学
光催化产氢装置
光催化构建双环吡唑啉和吡唑结构
光化学反应仪进口风险
光催化N-苯基哌啶β-C(sp³)–H酰胺化反应
烯烃的反马氏氢硫化反应
光诱导Co催化还原不对称交叉偶联
连续流微反应器
室温光化学反应仪
玻璃连续流反应器
太阳能驱动耦合催化CO2还原为合成气
光催化CO2还原制乙醇
石英微通道光催化反应器
光去羰基反应方程式
多功能光化学反应仪功能
光氧化还原催化重排级联反应
可见光诱导甲酸盐还原脱卤环化合成吲哚酮类化合物
光诱导硝基氧化合成
光反应釜搅拌方式
光化学反应仪配套设备
多通道光催化反应器
多功能光化学反应仪器
光催化剂制备过程
光化学
光催化烯基卤与α-硅胺的交叉偶联构建烯丙基叔胺
光催化制备前-芳香中间体
可见光化学反应仪
光催化构建氢噻吩和吡咯结构
硝基苯乙炔修饰
光催化合成C₂H₆
地屈孕酮制备设备
可见光诱导催化卤代吡啶
脂肪族异硫氰酸酯
寡聚物
光催化芳烃的氟磺酰甲基化
光催化氧化甲烷
光诱导钯催化体系
平行光反应仪技术参数
复旦大学张立武
多功能光化学反应仪
光催化芳基醚C−H氧化
板式微通道反应器
光化学羰基自由基生成方法
可见光诱导唑类化合物C-H芳基化反应
光催化合成吡唑硼化物
Nature
撬装微通道反应器
光催化制氢产率
光化学反应仪操作指南
光催化甲烷转化
光催化降解水中有机污染物
光化学衍生装置
光催化降解氟喹诺酮类抗生素
光催化N-酰胺
光氧化还原催化烯烃
光电催化
微通道反应器材质
上海光化学反应釜
高通量光反应仪
光化学反应机理
光催化胺烷基化
光催化甲烷制乙醛
光化学反应仪价格
光催化芳基卤和羧酸构建硫酯
led光催化反应器
光催化胺烷基化反应
光催化磺酰胺氮C-H键芳杂环反应
光化学led光源波长
光催化醛交叉偶联构建手性α-醇酮
曼彻斯特大学
光氧化还原实现消旋联烯与醛还原偶联反应
脂肪族羧酸脱羧卤代
光催化N-糖苷合成
光催化还原
光催化甲烷
可见光催化合成三环氮杂芳烃
光催化合成烯丙基砜衍生物
华中科技大学
光诱导γ-杂芳基化修饰新方法
光化学反应仪结构特点
光驱动耦合催化CO2还原为合成气
光催化二氧化碳还原制乙烯
全波段光化学反应仪
可换光源
光催化制苯乙烯
烯烃的氢甲基化反应
光电催化硝酸盐还原产氨
光催化碳碳偶联反应
光催化合成磺酰氟硼化物
光催化实验设备
光催化α-羟基酸
光诱导构建杂环或双环化合物
自由基还原交叉偶联
光氧化还原催化
光催化制2,5-二甲酰基呋喃
平行光反应仪
光催化烷烃与芳基溴化物交叉偶联
连续流光反应釜应用场景
光引发的偶联反应
大容量光化学反应仪
光催化芳基溴的选择性氰甲基化
上海大学
光化学反应仪地区分布
光催化脱氢乙烷制乙烯
光催化合成氮杂环丁烷化合物
光催化降解BPA
光催化乙炔制乙烯
有机光氧化还原催化剂10-苯基吩噻嗪
光催化有机材料合成
连续流光反应器
led光反应器
光催化甲烷制甲醛
光催化有机污染物矿化
釜式光反应器厂家
光反应器
武汉光化学反应仪厂家
光化学反应光源
光催化共轭二烯的对映选择性碳胺化反应
光催化醇脱氧芳基化
光化学反应仪化工领域应用
光催化甲烷制乙烷
光化学反应方程式
光催化制苯甲酸
光催化三氟甲基亚磺酸钠
LED光反应仪
光溴化反应釜
光催化光源
地屈孕酮工艺流程
光化学合成维生素B
光催化氧化自由基极性交叉
光谱学技术化学反应机理研究
光催化环加成反应构建碳环
低温光化学反应仪
光催化合成合成2,3-二胺化合物
风冷平行光反应仪
可见光催化葡萄糖
可见光催化环己烯制环己烯酮
国产光化学反应仪厂家
芳香酮化合物光敏剂
光催化合成2-取代萘
光催化脱羧C–C交叉偶联构建偕-二硼化合物
光催化制备2,5-己二酮
风冷控温平行光反应仪
双原子催化CO2光合成C2H4
国产光化学反应仪公司
普林斯顿大学
光催化γ-氨基官能化修饰
工业级光流体反应器
光化学反应仪参数
光化学合成硫醚
光催化反应基本过程
可见光诱导双核金催化脱卤硼化
光催化合成2,3-二氢苯并吡喃-4-酮并吡咯烷类化合物
分子光谱学应用
光催化还原制酮
可见光催化不对称烯丙基烷基化反应
光催化苯酚
光催化吡啶重排环化
光催化氧化甲苯
光催化合成应用
光催化制酯
光催化实现烯烃的烷氧基重氮甲基化
光反应器LED光源
可见光催化还原芳香硝基化合物
光催化重排合成芳乙胺
内照式光化学反应釜
光诱导重排环化
高通量光反应仪选型
可见光催化氧化伯、仲苄基 C(sp3)-H 键的亲核胺化
异噻唑啉酮光催化降解
光催化反应器常见故障解决办法
光催化接力不对称催化
微通道反应器的材质选择
光化学合成β-氨基醇
电化学
光催化自由基诱导碳碳双键和官能团易位
上海光化学反应仪
光去羰基反应原理
bromide
光催化空气净化
光催化丙烯醛自由加成反应
光催化实现苯环对位C-H键胺化
6000ml连续流光反应器
二氟烷基自由基加成
光诱导构建硫代酰胺
连续可见光催化
光催化sp3C−H键氧化反应
光催化尾气分解
高压光化学反应釜厂家
光催化合成α-硼基醛
玻璃光催化反应釜耐压选型
光催化烯烃异构化
金属光氧化还原交叉偶联
光催化反应基本原理
光催化磺胺嘧啶降解
光促进脂肪胺远程C(sp3)–H键溴化
光催化反应器波长筛选
多位光化学反应仪
光化学反应特点
臭氧氧化
常州光化学反应仪厂家
地屈孕酮光合成设备
光催化甲苯氧化
光催化合成烯烃
光催化反应釜常见故障问题
液相光化学反应釜
实验室级光化学反应仪
微通道反应器定制
中山大学
天津光化学反应仪
光催化蒽的不对称[4+2]去芳构化
光催化醇与氯代芳烃的脱氧交叉偶联反应
JACS
串联LED光反应仪
常温常压光解塑料
光催化芳基环丙烷和硝酮偶极环加成反应
光催化降解
光催化CH4
可见光催化氧化脱氢
光流体反应器
光催化对映选择性C-H官能团化实现吲哚的不对称去芳构化
可见光催化芳香杂环氮自由基对非活化烯烃加成反应
光催化反应釜原理
小试级光反应器
深紫外光反应器
可见光诱导的脱羧烯丙基化反应
江苏大学
单工位全波段光反应仪
光催化H2O2
光催化光敏药物合成
光诱导Pd催化烯丙基C-H氧化
光催化烷基C-H键选择性末端硼化转化
光催化构建糖胺
光催化烯基重氮[3+2]环加成反应
光催化对烯烃进行烷氧基化重氮甲基化
可见光催化活化C-H氨基化的反应方法学
中国科学院
可见光催化芳烃C-H胺基化
光催化合成反应釜
光流体微反应器
芳基环丙烷与硝酮环加成反应
光催化甲烷转化为高附加值化学品
可见光催化N-烷基化
光化学反应仪分类
N-烷基苯胺
光催化合成苯并膦氧化合物
光催化甲烷制甲醇
光诱导催化sp3C−H键卤代
深紫外光化学反应仪厂家
吉林大学
微通道反应器持液量
光催化氧化还原反应
连续流光反应器
光诱导催化偕二氯烷烃的发散性去氯硼化反应
并联LED光反应仪
光催化合成氨基酸
光化学反应仪生产厂家
平行光反应仪维护
光催化对硝基苯
光催化制醛
光催化环丙基酮去消旋化
光反应器光源
光诱导Pd催化制备芳基自由基前体
安徽大学
光催化脱硫反应釜
光异构化
光催化合成多取代氮杂环庚烷
紫外光化学反应仪
可见光光催化分子氧活化
光催化制H2O2
光催化医药合成
光催化反应器使用说明
光致异构化合成应用
北京光化学反应仪报价
陕西科技大学
光诱导镍催化烯烃碳卤化反应
可见光催化二芳基醚的C-O键断裂
光诱导合成瓶刷聚合物
光催化醚的脱氧交叉偶联
高压光化学反应釜报价
可见光光氧化还原
石英玻璃反应釜
PHECOO
光催化和钴催化非活性烯烃环异构化合成杂环
光化学反应仪使用说明
光催化烯烃碳卤化反应
光催化蒽醌基环三核铜配合物
羧化反应
光催化合成烯丙基硅化合物
光流体微通道反应器选型
光合成
风冷LED光反应仪
工业级连续流反应器
光催化析氢反应器
光催化芳基溴的选择性氰化
郑州光化学反应仪报价
拉曼区光化学反应仪
光化学反应仪光强分析
光催化污水处理



